Theoretical Biophysics Lab
京都大学理学研究科 生物物理学系
理論生物物理学分科

-
募集
大学院生・研究員募集中
Theoretical Biophysics Lab
京都大学理学研究科 生物物理学系
理論生物物理学分科
大学院生・研究員募集中

1965年生まれ。1988年京都大学理学部卒、1990年同大学院理学研究科化学専攻修士修了、 1991年~1995年岡崎国立共同研究機構技官(分子科学研究所)、1995年~1998年日本学術振興会研究員(イリノイ大学化学科)、 1998年~2001年神戸大学理学部化学科講師を経て、2001年より神戸大学理学部化学科助教授、 2007年より京都大学理学研究科生物科学専攻生物物理教室准教授、2013年より、同教授。総合研究大学院大学博士(理学)。
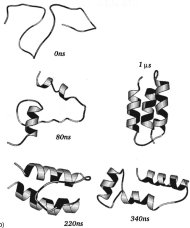
p53は有名な癌抑制作用のある蛋白質です。分子生物学的には、p53は転写制御因子として働きます。すなわち、ゲノムに存在する数万の遺伝子の中からそのとき必要な遺伝子を発現するために、その遺伝子の制御領域にp53などの転写制御因子が結合し発現を促します。私たちは、粗視化分子シミュレーションによって、p53がゲノム上でターゲット遺伝子を探して動き回る様子と、ターゲット遺伝子のp53結合領域(応答エレメント)を見つけてそこに留まる様子を解析しました。前者の動き回る様子は、Terakawa et al JACS 2012で報告し(図の左)、後者の応答エレメントを見つける様子を本論文で報告しました(図の右)。面白いのは、まず、p53が極めて柔らかい分子で、全体として常に構造を変えながら動いていることです。さらに、動き回るときと見つけて留まるときとで、全く異なる形をしています:動き回るときは、C末端の赤い部分でDNA上を滑るようにスライディングしますが、留まるときには緑色のコアドメインが応答エレメントを認識して強く結合します。このような動態を明確に示すことができるのが分子シミュレーションンの特色と言えるでしょう。
p53についての研究は、寺川君が大学院修士と博士課程で一貫して研究したものです。最初は、p53が著名な癌抑制因子ということで創薬などを念頭においてこの蛋白質を研究することになってのですが、次第にこの蛋白質自身の面白さに引き込まれていったように思います。天然変性領域をもつために大きくゆらぎ、高次構造を変えること、一つの分子内に2か所のDNA結合領域があること、さまざまな翻訳後修飾を受けること等々、多様な面白さが詰まっています。研究室としても、p53の研究をきっかけに、研究テーマを遺伝子動態解析にシフトしていくことになりました。
転写因子p53のDNA上のスライディングによる遺伝子探索過程のムービーはこちらで、p53の胴体についてはこちらで美しい映像を観ることができます。
生体分子シミュレーションというと、主流は原子モデルを用いた分子力場による分子動力学シミュレーションです。しかし、この方法で計算できる時間スケールは現在のところマイクロ秒程度であり、これは多くの生命現象に要する時間より桁違いに短いのです。私たちは、より生命らしい現象を扱うためにはより長時間のシミュレーションが不可欠であり、そのために原子レベルよりも分解能を粗視化したモデルによるシミュレーションが有効だと考えています。そこで、粗視化モデルの構築を進めるともに、それを具現化するものとしてソフトウエアを開発してきました。そのソフトウエアCafeMolを解説したのが、この論文です。
過去10年以上にわたって進めてきた粗視化モデルによるシミュレーション法を、他の研究者にも使ってもらいたい、ということでソフトウエアを公開しています。興味のある方はぜひ、ダウンロードして、ご利用ください。
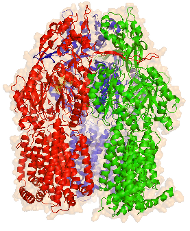
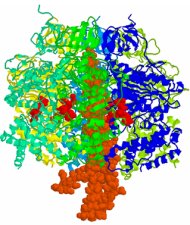
多剤耐性は、院内感染やがん化学治療など社会問題を引き起こしています。緑膿菌などの多剤耐性化の主な原因になっているのはRND型の多剤排出トランスポーターの発現量増加です。大腸菌のRND型多剤排出トランスポーターであるAcrBは、村上聡らによってX線構造解析がなされ、それに基づいて機能的回転機構という作動機構が提唱されています。この論文は、私たちが開発してきた粗視化分子シミュレーション技法を応用して、AcrBの薬剤排出過程のシミュレーションを行い、それに基づいて機能的回転機構を計算機上で実現したものです。計算機実験では、プロトン結合に起因する薬剤の解離過程が全体のサイクルのボトルネックになっていることが示唆されました。
この研究は、次世代スパコン「京」の有効活用のためのソフトウエア開発の一環として行われました。2008年ごろから、村上さんとの共同研究で、分子スケールのさまざまなレベルの計算研究が同時に進められてきました。この論文はそのなかの第一弾として、粗視化シミュレーションによって薬剤排出過程を調べた研究成果です。次世代スパコン「京」により、原子レベルのシミュレーションが加速し、もっと大きな成果になることを期待しています。
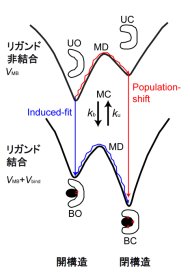
相手分子との結合に伴う蛋白質の構造変化は古くて新しい問題です。伝統的にはKoshlandの誘導適合がもっともよく受入れられていますが、最近はMWCにその起源をもつpopulation-shiftモデルが(再?)注目されています。Okazaki et al 2006で提案した粗視化モデル=多谷モデルを拡張したシミュレーション技法で、誘導適合vs population-shiftの問題を調べたのがこの論文です。私達の計算では、結合に伴う構造変化において誘導適合とpopulation-shiftは並列の経路であり、どちらをどれくらい通るのかはケース依存であるということになりました。特に、リガンドと蛋白質の相互作用が弱いとき、および短距離力のときはpopulation-shiftが支配的であり、反対に相互作用が強く、長距離力のときは誘導適合が支配的になります。従って、蛋白質と薬剤などの小分子の場合はpopulation-shiftが優勢で、蛋白質と核酸の結合では誘導適合が優勢になるはず、と主張しています。
これ、理由(種明かし)を読むと、自然というか、もっと言えば当たり前というか、そんな印象を受けるかもしれません(そんな感想を何回か聞きました)。しかし、私達が言うまで誰もこんなことは言ってなかったのだから、そんなに自明なことではないのでは、、と言っています。実験的なサポートは、少しありますが、それほど多くはありません。この結論が実際の蛋白質にどれくらい適用できるのか、今後徐々に明らかになってくればいいなあと思っています
回転分子モーターF1-ATPase(図)は、その回転が日本の一分子実験で実証された有名なシステムです。最小回転単位はアミノ酸~3000個程度を含み、ミリ秒オーダーの時間で回転します。原子レベルの分子動力学計算で、分子モーターとしての機能を直接追うことは到底不可能です。私たちは、アミノ酸を一つの球で近似した郷モデルをスイッチされるというアイデアでATPの効果を模倣することによって、回転運動のシミュレーションを実現しました。このセットアップを使って多数の化学力学共役の経路を試した結果、実験結果と無矛盾な経路はalways-bi-siteモデルであることを主張しました。
この研究は、2001年夏に野地さんの講演を聞いて興味をもったことからスタートしました。その年の秋には、古賀君はF1を計算機の中で回転させることに成功していました。初めて回るムービーを観たときは、非常に感激しました。それが、、、、続々と出てくる一分子実験の結果に翻弄されながら、最終的に論文出版は2006年になってしまいました。最後は、古賀君の粘り強さでalways-bi-siteモデルに行き着きました。2007年Cellに出た一分子実験は、ほぼこのモデルになっています。
高田研としても、この研究をきっかけに、研究の舵を分子モーターの方に大きく切っていくことになりました。その意味でも非常に貴重な論文です。
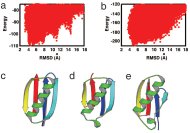
蛋白質立体構造予測の成否の知見からフォールディング原理を学ぼうと言う論文です。構造予測の研究分野で、フラグメントアセンブリ(FA)法というのが今のところもっとも有力です。なぜFA法はそんなにうまく機能するのかを、千見寺君が探求した結果です。局所構造には対象蛋白質の配列情報を使い、非局所相互作用には配列に寄らない力だけを用いたキメラシミュレーションを行いました。物理屋らしい千見寺君のアイデアです。キメラ計算でも、対象蛋白質の立体構造が出てくるところが見せ場(図a,dが本来の計算、b,eがキメラ計算によります。キメラ計算の予測結果eは、少しゆがんでいるけど、天然構造トポロジー(図c)を持っています)。
なかなか美しい論文だと思っています。内容的には、アイデアも含めて、ほぼ千見寺君の仕事です。
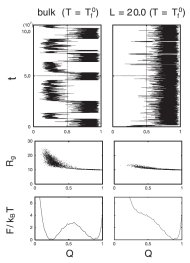
分子シャペロンの代表格シャペロニンが、いかにして蛋白質フォールディングを助けるのかという問題に関して、”箱の中に閉じ込めることで蛋白質を安定にし、フォールディングを加速できること(ただし箱が狭すぎると逆に減速する)を示唆したシミュレーション研究です。Hartlらが実験的に示唆したことに刺激されて、郷モデルで計算してみたという内容です。理論的にはシンプルな話ですが、実験の方から反響がわりと大きかった。図の左が箱の無いとき、右が箱の中でのシミュレーション。箱が無いときには許されていた、伸びた変性状態構造が、箱の中では許されない(中段)ので、箱の中では変性状態の自由エネルギーが高くなる(下段)、ということです。
後に、Hartlらは2006年のCell論文で、私たちが予測したフォールディング速度の加速→減速という関係を、実験的に検証してくれました。
蛋白質デザインに関する論文です。コンピュータで全長配列をデザインした人工蛋白質を実験グループ(神戸大・田村研)が合成・物性測定してくれて、テストしたものの中の一つがちゃんと三次構造をとることを確かめた、という仕事です。とくに、デザイン原理として、蛋白質のエネルギー地形をファネル状にすることも目的関数とした点は、かなりユニークな仕事です。この後もこの方向でβ蛋白質のデザインを試みましたが、ずっと失敗続きでした。
高田研として実験とうまくコラボした貴重な経験でした。出版に相当苦労したけど、最後はStructureの表紙を飾れて、ほっとした!
複雑系のサンプリング手法として今では広く普及したレプリカ交換法についての論文です。この時までほとんどの場合、温度が異なるレプリカが使われていたけど、ハミルトニアンを変えたレプリカ交換法はより一般的で便利だよ、という提案でした。内容的にはそれほど高度なものではありませんが、ここで名づけたHamiltonian replica exchange methodという用語は業界で定着し、現在もいろいろ発展しています。
高田研としては稀有な、計算手法の開発に関する論文です。そのあたりのテレがあって、タイトルがなぜかOnで始まっています。

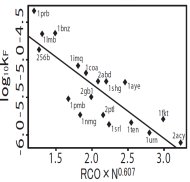
非格子型郷モデルを使って、蛋白質フォールディング経路と速度をサーベイしました。古賀君が自作したプログラムをうまく自動化した結果、あれよあれよと言う間に18個の蛋白質で調べ上げられ、図のようなきれいな関係を見出しました。フォールディング速度が、構造トポロジーパラメータRCOと長さの約1/3乗でスケーリングするという結論は、この後フォールディング反応研究の分野で随分議論されることになりました。
神戸で研究室を立ち上げその学生が第一著者になった最初の論文で、思い出深いものです。これはほぼ古賀君の4年生の卒研研究の成果です。comparative MDと言う研究手法、つまり網羅的に計算機実験して”比較”すると大事なことが分かるんだなあ、ということをこの論文で知りました。その後の研究姿勢に大きな影響を与えています。
蛋白質フォールディング過程をシミュレーションするために、ある程度物理化学的な粗視化モデルを作り、3本へリックスバンドルの人工配列ペプチドに関してフォールディングシミュレーション(図は、トラジェクトリのスナップショット)を行った論文です。
これからずっと開発を続けているソフトウエアSimFoldを使った最初の論文です。米国ポスドク時代の96年ごろから研究を始め、神戸大に移ってからも相当苦戦した思い出の仕事です。とくに、乱数アルゴリズムがまずかったせいで、半年以上無駄にしました。それが分かったときのショックが忘れられません。